COMING TO GRIPS WITH LYMPHOCYTIC THROMBOPHILIC ARTERITIS

By Warren R. Heymann, MD, FAAD
May 18, 2022
Vol. 4, No. 20

In 2003, Fein et al described 3 cases of what they termed macular arteritis (MA) — a novel form of cutaneous arteritis in which the primary lesion is a hyperpigmented macule, without any classical features of cutaneous vasculitis such as palpable purpura, erythematous nodules, or ulceration. Their patients (all African American; 2 women and 1 man) were asymptomatic and followed an indolent course for periods of 2 to 15 years. Systemic involvement was not observed. Histology demonstrated a marked lymphoplasmacytic inflammatory infiltrate centered around a small artery. The ESR was elevated in 2 patients; 2 had weakly positive anticardiolipin antibodies; ANCA was negative in all 3 patients. (2)
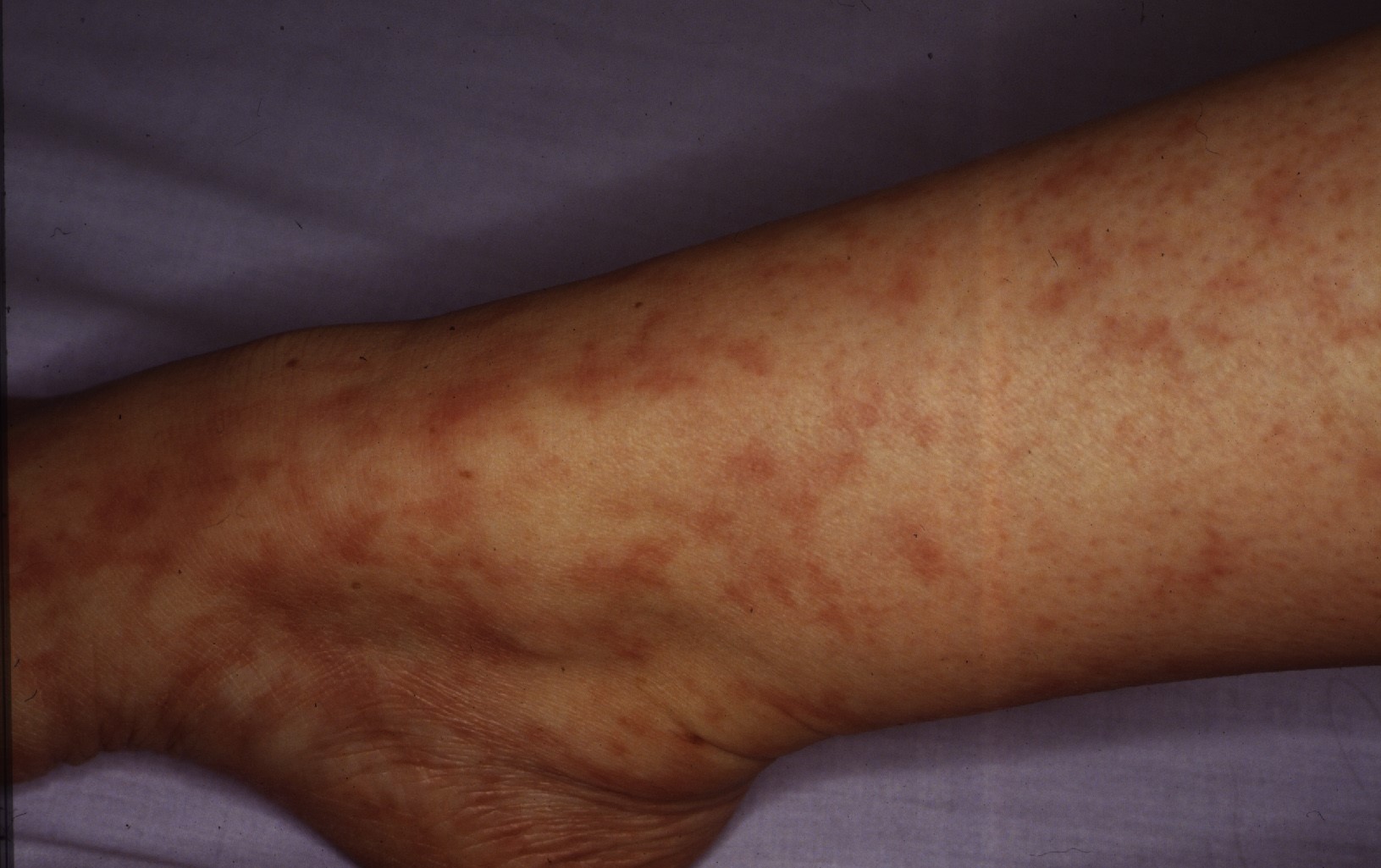
Five years later, Lee et al described 5 women (mean age, 25 years; age range, 20-34 years) with persistent, slowly progressive, patchy and reticular hyperpigmentation associated with livedo racemose, predominantly affecting the lower extremities. Most patients had only "subtle palpable subcutaneous indurations, with 1 patient having more prominent nodularity and papules." Biopsies revealed infiltration of the muscular vessel wall by inflammatory cells, affecting small arteries of the dermosubcutaneous junction or superficial subcutis. At least 90% of the infiltrate were lymphocytes with an admixture of histiocytes. Neutrophils and eosinophils were absent or scant, although one image clearly demonstrates intraluminal nuclear dust. A concentric fibrin ring involving the entire periphery of the lumina of affected vessels was present in all the patients, reminiscent of a thrombophilic state (which the authors thought could represent a localized thrombophilia triggered by lymphocytic endovasculitis). Laboratory studies demonstrated serum antiphospholipid antibodies in 4 patients, although none fulfilled criteria for having the antiphospholipid syndrome. One of these patients had a heterozygous mutation of the factor V Leiden gene. The authors believed that the vasculitic process in their patients was identical to those with MA as described by Fein et al. The authors concluded that the term "lymphocytic thrombophilic arteritis" best reflected the histologic features combining lymphocytic vascular inflammation with changes representing a thrombophilic endovasculitis. (3)
In an accompanying editorial, LeBoit opines: "Whether one calls this emerging condition macular arteritis (which invites confusion with a retinal condition) or lymphocytic thrombophilic arteritis, as Lee et al propose, it seems likely that there is now a condition in which lymphocytic vasculitis, in this case arteriolitis, is a characteristic finding." (4)
Macarenco et al reported 2 cases of biopsy-proven cutaneous lymphocytic arteritis. A 19-year-old white woman presented with hyperpigmented macules (and a few erythematous macules and papules) of her legs, that had been mildly painful a few months earlier. Other than an elevated ESR, her serologic evaluation was normal. The patient declined treatment and her lesions persisted over the next year. The second patient was a 26-year-old woman, with a prior history of deep vein thrombosis (with a heterozygous prothrombin mutation and wild type Factor V Leiden and plasminogen activator type-1 genes) presented with a 1-week history of leg pain and swelling, rash, fever, and pleuritic and epigastric pain. Serologic studies were negative, as were her Doppler and chest CT studies. A prompt remission was observed with a prednisone taper. Based on these cases and their literature review, the authors suggested that MA, LTA, and CPAN represent a spectrum of illness: "MA and LTA can be considered latent, non-nodule forming variants of CPAN [cutaneous polyarteritis nodosa], exhibiting less severe inflammatory destruction of the vessel wall and less pronounced neoangiogenesis (a histologic correlate with nodule formation). Increasing disease severity moving from MA to LTA to CPAN to systemic PAN is likely manifested by the accumulation of more numerous and varied types of vasculitic lesions (e.g., nodules, ulcers, foot drop) and acquisition of autoimmune serology (eg, ANA, RF, APA, and/or ANCA)." (5) In their systematic review of LTA, Vakili et al propose that MA and LTA are identical entities, and suggest that just the term LTA be utilized in instead of MA or MLA. (6)
Therapy for LTA has been mostly supportive, as no cases have been reported to progress to systemic vasculitis. Corticosteroids (topical or systemic), anticoagulants, antibiotics, and narrow band UVB have been utilized with little or no benefit. (7)
To date, there have now been approximately 50 reported cases of LTA and the controversy as to whether this entity is part of the CPAN spectrum remains unsettled. Buffiere-Morgado et al performed a retrospective study of 35 patients to assess the frequency of clinical and histologic features of MLA in patients with cPAN. All 35 patients had an infiltrated livedo, nodules, or both. Ulceration was rare. Erythematous or pigmented lesions were present in 54% of patients. Predominantly lymphocytic arteritis, a paucity of neutrophils, concentric fibrin ring, and absence of internal lamina elastic disruption were present in 60%, 20%, 18%, and 23% of patients, respectively. Median follow-up was 11 years. None of the patients had systemic involvement, and 57% had a complete remission. The incidence of complete remission was not different between patients having a predominant lymphocyte infiltrate or few neutrophils. The authors concluded that their data do not favor the classification of CPAN and MLA as distinct entities. (8) Other authors disagree. Kelly et al performed a cross-sectional study of 17 patients with LTA and 13 patients with CPAN. "Clinically, cases of LTA were distinguished by a more widespread pattern of livedo racemosa, which was non-infiltrated and asymptomatic. In contrast, cPAN was associated with localized starburst livedo, purpura, and episodic features including nodules, pain, and large inflammatory ulcers. When patients were separated according to the presence (>5%) or paucity (<5%) of neutrophils on blinded histology review, they had distinct clinical features and differences in disease course." The authors assert that their data supports the classification of LTA and CPAN as separate entities, underscoring the importance of clinical-pathologic correlation in distinguishing these disorders. (9)
In my estimation, the quandary of whether LTA falls within the spectrum of CPAN will not be answerable until we completely understand the pathogenesis of each disorder. Regardless, there is now ample literature to convince me that lymphocytic vasculitis is rare but real. Until such time when authorities definitively comprehend the etiology and nosology of LTA, the most prudent approach is to reassure the patient that this is likely a benign process, with an excellent prognosis, while following the patient carefully, being certain that there is no systemic vasculitis.
Point to Remember: Lymphocytic thrombocytic vasculitis is a rare disorder with a good prognosis. Whether this falls under the spectrum of cutaneous polyarteritis nodosa, or is a disease sui generis, will be determined when the etiology of these diseases is ascertained.
Our expert's viewpoint
Jason B. Lee, MD, FAAD
Clinical Vice Chair
Director, Jefferson Dermatopathology Center
Director, Residency and Dermatopathology Fellowship
Director, Jefferson Pigmented Lesion Clinic
Jefferson University Hospitals
Whether one believes macular arteritis and the more recent designation lymphocytic thrombophilic arteritis (LTA) is a distinct entity or it represents a spectrum of cutaneous polyarteritis nodosa (cPAN) will depend on which body of evidence one chooses to believe, as there is literature that supports either position. The rarity of both diseases makes it difficult to have a firm conviction about their nature. One must rely on limited or non-existent experience with the diseases and the scant body of evidence in the literature.
Those who believe that LTA is a distinct entity focus on the clinical and histopathological differences when compared to cPAN, a view shared by LeBoit in his editorial where he affirms the existence of lymphocytic vasculitis pointing out that LTA is a prime example of one. (2-4, 9) Those who have a contrary view argue that the clinical and histopathological findings are not sufficiently distinct and that they can be encountered in the spectrum of clinical and histopathological presentation of cPAN, lumping the rarer LTA into a more established disease. According to these authors, the predominance of lymphocytes in cases reported as LTA represents late or reparative stage of cPAN. (5,8)
Neither point of view is supported by strong scientific body evidence, supported only by case reports and limited number of small studies. First is the issue of lymphocytic vasculitis, that is, whether lymphocytes may serve as the primary mediator of vasculitis. While neutrophilic vasculitis is well-established accounting for a majority of the vasculitis encountered in the skin, lymphocytic vasculitis is exceedingly rare. There is even debate whether this type of vasculitis even exists at all, some asserting that it merely represents an epiphenomenon or a secondary finding and not part of a primary pathologic process. I am part of the group who share this view for I have yet to encounter a convincing case of lymphocytic vasculitis as a dermatopathologist. In the few instances over the last 25 years where I have encountered the histopathologic changes described in LTA, I have signed them out as medium vessel vasculitis consistent with or favoring polyarteritis nodosa. Clinically, they were confirmed to be either a thrombotic vasculopathy of hypercoagulable state or cPAN. From my vantage, there is insufficient evidence to conclude that LTA is a clinically and histologically distinct entity. Late-stage or subacute forms of cPAN may account for the clinical and histopathological findings described in LTA. Until more convincing evidence emerges, I consider LTA as either a subset of cPAN or thrombotic vasculopathy rather than a disease sui generis.
All content found on Dermatology World Insights and Inquiries, including: text, images, video, audio, or other formats, were created for informational purposes only. The content represents the opinions of the authors and should not be interpreted as the official AAD position on any topic addressed. It is not intended to be a substitute for professional medical advice, diagnosis, or treatment.
DW Insights and Inquiries archive
Explore hundreds of Dermatology World Insights and Inquiriesarticles by clinical area, specific condition, or medical journal source.
Skin Care Physicians of Costa Rica
Clinica Victoria en San Pedro: 4000-1054
Momentum Escazu: 2101-9574
Please excuse the shortness of this message, as it has been sent from
a mobile device.
posted by dermatica at
May 18, 2022
![]()
![]()

0 Comments:
Post a Comment
Subscribe to Post Comments [Atom]
<< Home